Overview
This vignette is an introduction to the usage of pareg. It estimates pathway enrichment scores by regressing differential expression p-values of all genes considered in an experiment on their membership to a set of biological pathways. These scores are computed using a regularized generalized linear model with LASSO and network regularization terms. The network regularization term is based on a pathway similarity matrix (e.g., defined by Jaccard similarity) and thus classifies this method as a modular enrichment analysis tool (Huang, Sherman, and Lempicki 2009).
Installation
if (!require("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("pareg")Load required packages
We start our analysis by loading the pareg package and other required libraries.
Introductory example
Generate pathway database
For the sake of this introductory example, we generate a synthetic pathway database with a pronounced clustering of pathways.
group_num <- 2
pathways_from_group <- 10
gene_groups <- purrr::map(seq(1, group_num), function(group_idx) {
glue::glue("g{group_idx}_gene_{seq_len(15)}")
})
genes_bg <- paste0("bg_gene_", seq(1, 50))
df_terms <- purrr::imap_dfr(
gene_groups,
function(current_gene_list, gene_list_idx) {
purrr::map_dfr(seq_len(pathways_from_group), function(pathway_idx) {
data.frame(
term = paste0("g", gene_list_idx, "_term_", pathway_idx),
gene = c(
sample(current_gene_list, 10, replace = FALSE),
sample(genes_bg, 10, replace = FALSE)
)
)
})
}
)
df_terms %>%
sample_n(5)## term gene
## 1 g1_term_9 g1_gene_12
## 2 g1_term_5 g1_gene_7
## 3 g2_term_2 g2_gene_2
## 4 g1_term_3 bg_gene_47
## 5 g1_term_8 g1_gene_1Term similarities
Before starting the actual enrichment estimation, we compute pairwise pathway similarities with pareg’s helper function.
mat_similarities <- compute_term_similarities(
df_terms,
similarity_function = jaccard
)
hist(mat_similarities, xlab = "Term similarity")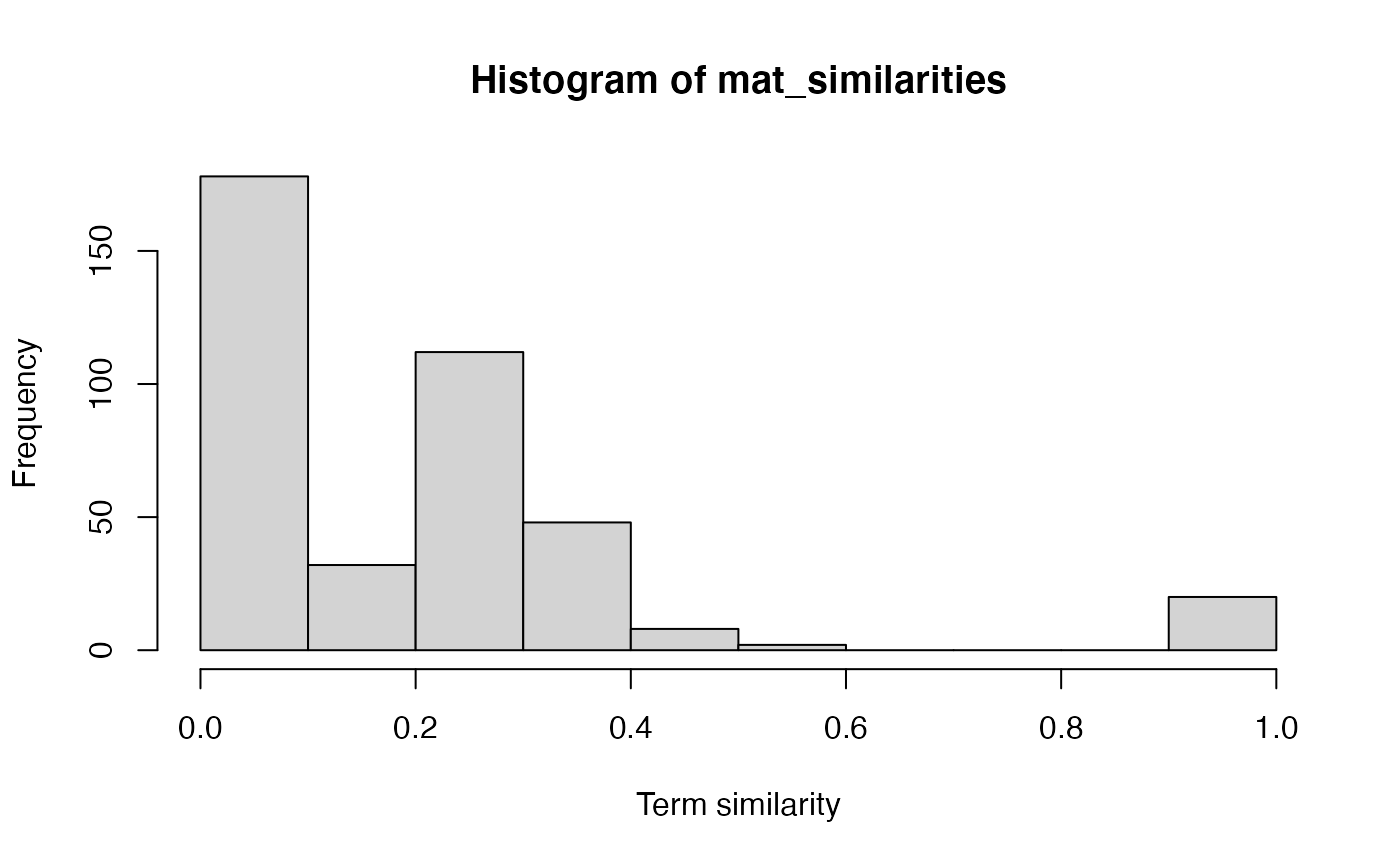
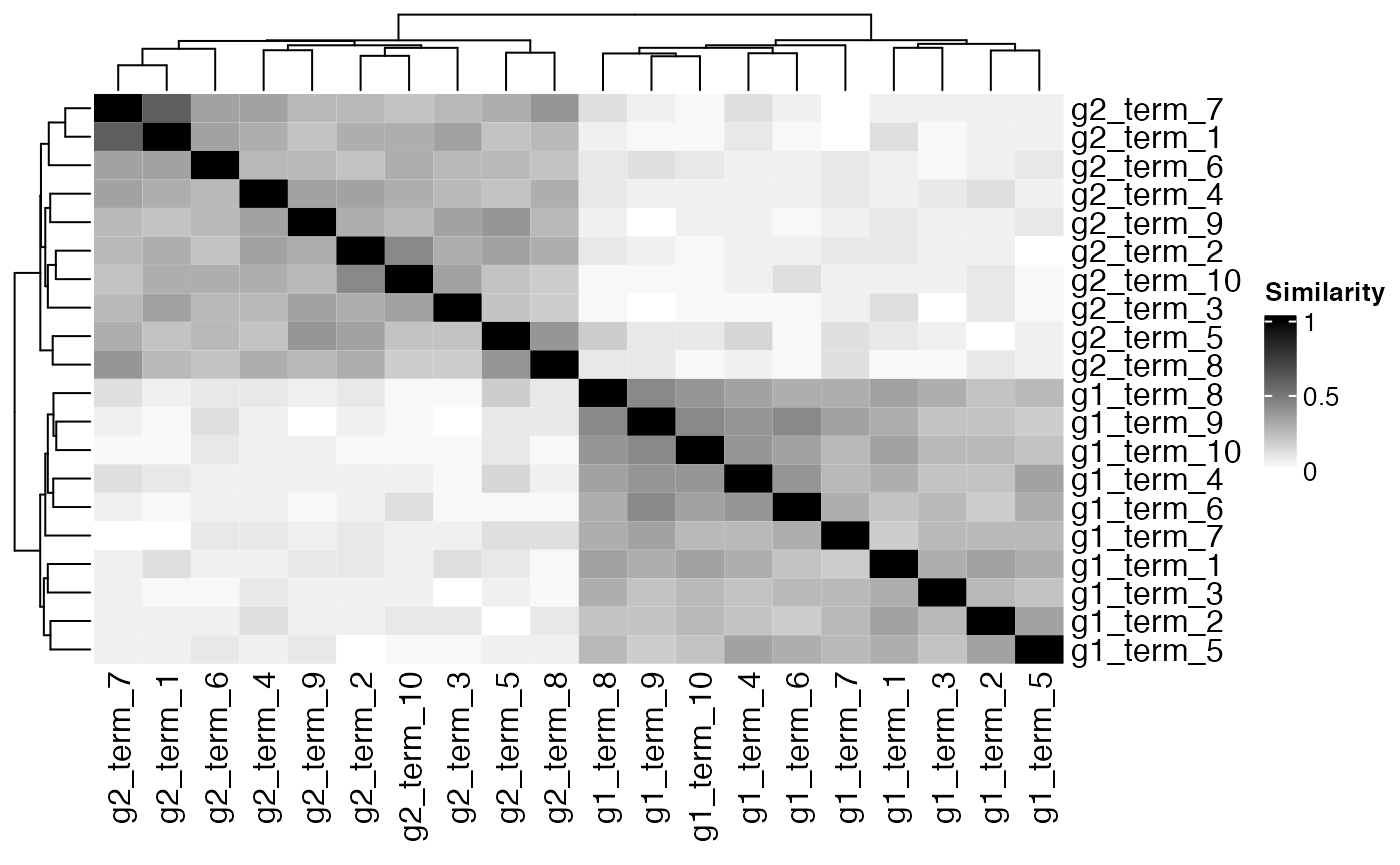
We can see a clear clustering of pathways.
Heatmap(
mat_similarities,
name = "Similarity",
col = circlize::colorRamp2(c(0, 1), c("white", "black"))
)
Create synthetic study
We then select a subset of pathways to be activated. In a performance evaluation, these would be considered to be true positives.
active_terms <- similarity_sample(mat_similarities, 5)
active_terms## [1] "g2_term_6" "g2_term_3" "g2_term_3" "g2_term_2" "g2_term_8"The genes contained in the union of active pathways are considered to be differentially expressed.
de_genes <- df_terms %>%
filter(term %in% active_terms) %>%
distinct(gene) %>%
pull(gene)
other_genes <- df_terms %>%
distinct(gene) %>%
pull(gene) %>%
setdiff(de_genes)The p-values of genes considered to be differentially expressed are sampled from a Beta distribution centered at \(0\). The p-values for all other genes are drawn from a Uniform distribution.
df_study <- data.frame(
gene = c(de_genes, other_genes),
pvalue = c(rbeta(length(de_genes), 0.1, 1), rbeta(length(other_genes), 1, 1)),
in_study = c(
rep(TRUE, length(de_genes)),
rep(FALSE, length(other_genes))
)
)
table(
df_study$pvalue <= 0.05,
df_study$in_study, dnn = c("sig. p-value", "in study")
)## in study
## sig. p-value FALSE TRUE
## FALSE 34 17
## TRUE 1 28Enrichment analysis
Finally, we compute pathway enrichment scores.
fit <- pareg(
df_study %>% select(gene, pvalue),
df_terms,
network_param = 1, term_network = mat_similarities
)## Loaded Tensorflow version 2.3.0The results can be exported to a dataframe for further processing…
| term | enrichment |
|---|---|
| g2_term_6 | -0.6760687 |
| g2_term_3 | -0.6005415 |
| g2_term_2 | -0.5818557 |
| g2_term_4 | -0.4233026 |
| g2_term_8 | -0.4123425 |
| g1_term_2 | 0.3978593 |
…and also visualized in a pathway network view.
plot(fit, min_similarity = 0.1)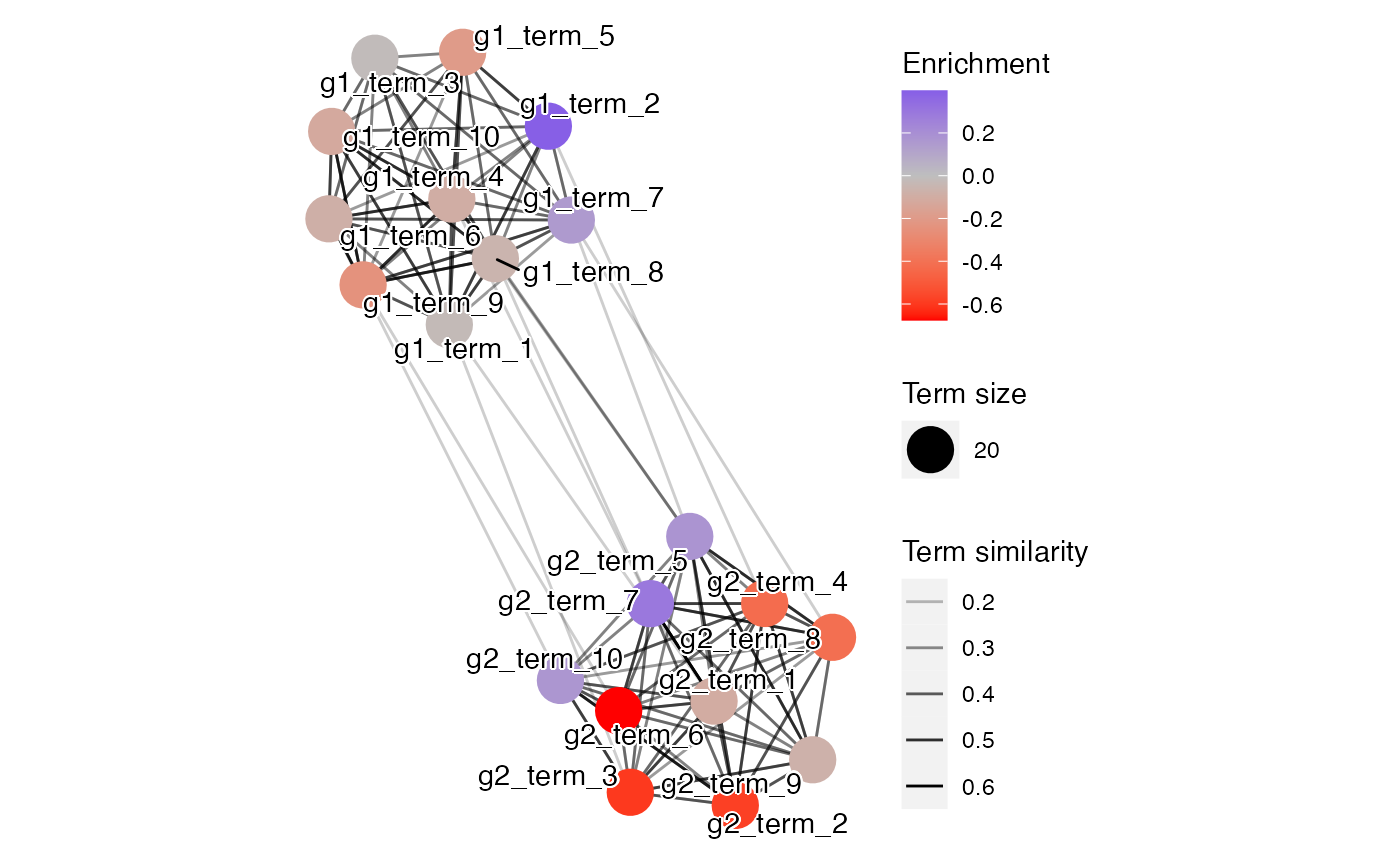
To provide a wider range of visualization options, the result can be transformed into an object which is understood by the functions of the enrichplot package.
obj <- as_enrichplot_object(fit)
dotplot(obj) +
scale_colour_continuous(name = "Enrichment Score")## Scale for 'colour' is already present. Adding another scale for 'colour',
## which will replace the existing scale.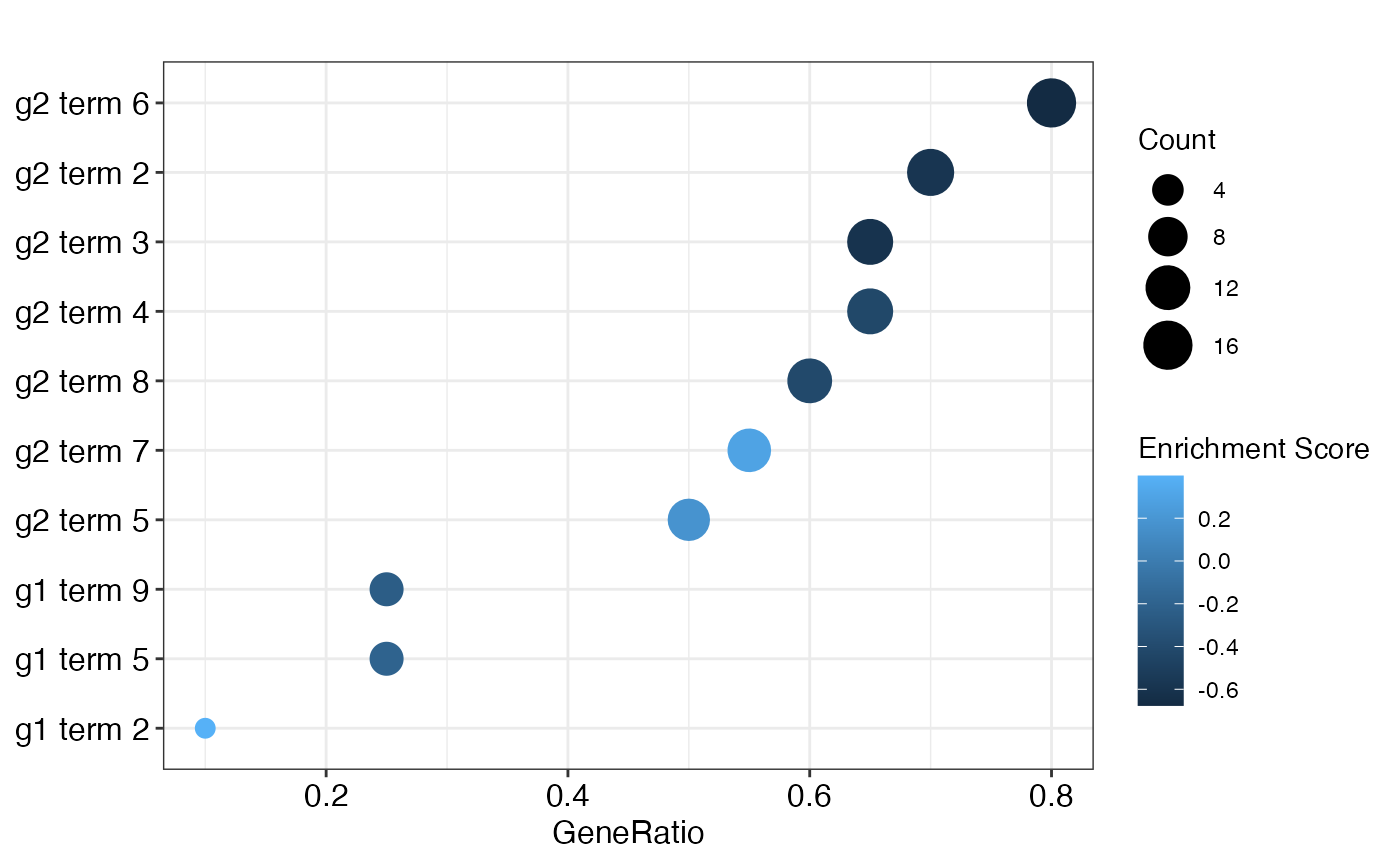
treeplot(obj) +
scale_colour_continuous(name = "Enrichment Score")## Scale for 'colour' is already present. Adding another scale for 'colour',
## which will replace the existing scale.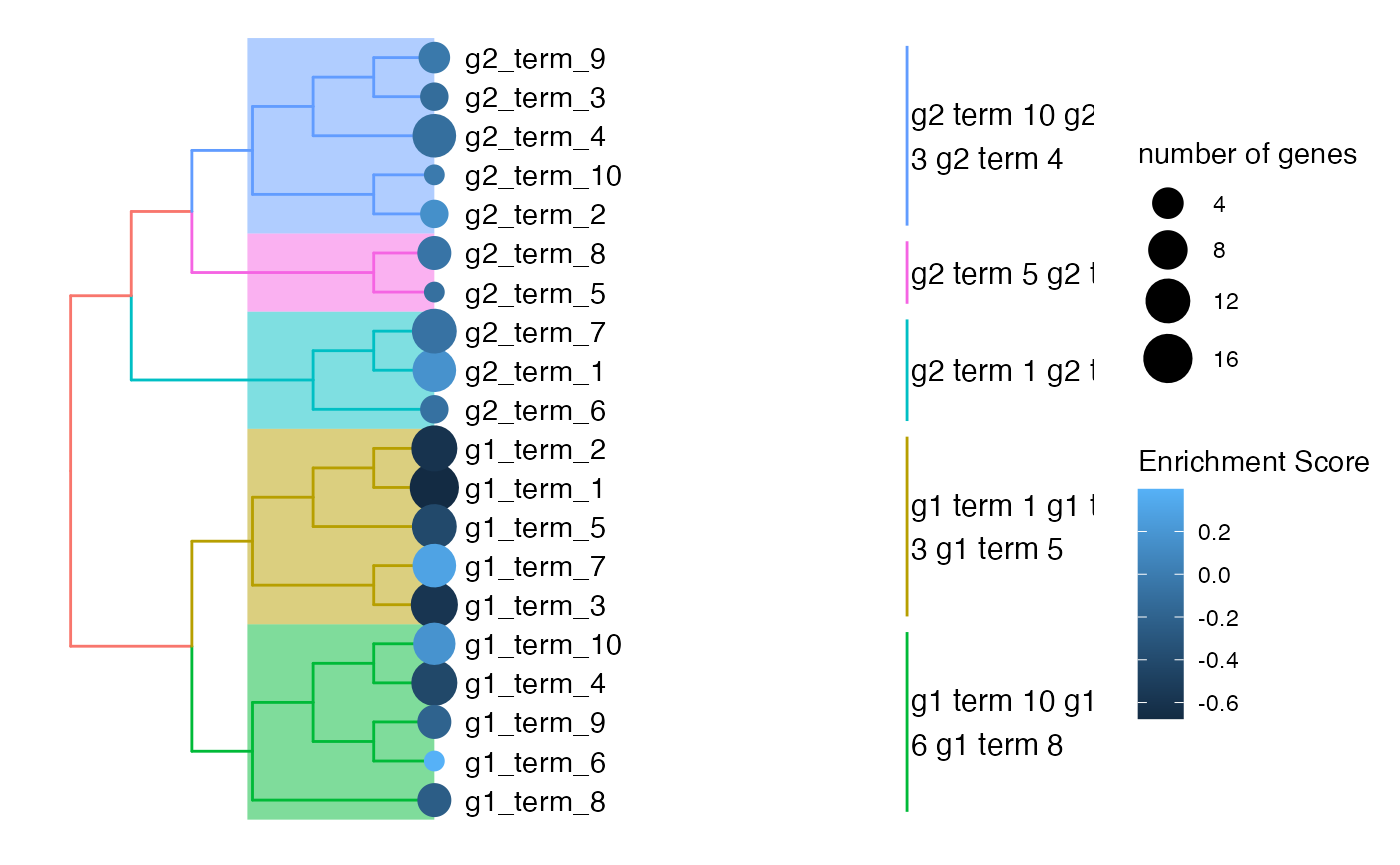
Session information
## R version 4.2.1 (2022-06-23)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur ... 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] grid stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] pareg_0.99.5 tfprobability_0.15.0 tensorflow_2.9.0
## [4] enrichplot_1.16.1 ComplexHeatmap_2.12.0 forcats_0.5.1
## [7] stringr_1.4.0 dplyr_1.0.9 purrr_0.3.4
## [10] readr_2.1.2 tidyr_1.2.0 tibble_3.1.7
## [13] tidyverse_1.3.1 ggraph_2.0.5 ggplot2_3.3.6
## [16] BiocStyle_2.24.0
##
## loaded via a namespace (and not attached):
## [1] utf8_1.2.2 reticulate_1.25-9000 tidyselect_1.1.2
## [4] RSQLite_2.2.14 AnnotationDbi_1.58.0 BiocParallel_1.30.3
## [7] devtools_2.4.3 scatterpie_0.1.7 munsell_0.5.0
## [10] codetools_0.2-18 ragg_1.2.2 future_1.26.1
## [13] withr_2.5.0 keras_2.9.0 colorspace_2.0-3
## [16] GOSemSim_2.22.0 Biobase_2.56.0 highr_0.9
## [19] logger_0.2.2 knitr_1.39 rstudioapi_0.13
## [22] stats4_4.2.1 DOSE_3.22.0 listenv_0.8.0
## [25] labeling_0.4.2 GenomeInfoDbData_1.2.8 polyclip_1.10-0
## [28] bit64_4.0.5 farver_2.1.0 rprojroot_2.0.3
## [31] parallelly_1.32.0 vctrs_0.4.1 treeio_1.20.0
## [34] generics_0.1.3 xfun_0.31 R6_2.5.1
## [37] doParallel_1.0.17 GenomeInfoDb_1.32.2 clue_0.3-61
## [40] graphlayouts_0.8.0 bitops_1.0-7 cachem_1.0.6
## [43] fgsea_1.22.0 gridGraphics_0.5-1 assertthat_0.2.1
## [46] scales_1.2.0 gtable_0.3.0 globals_0.15.1
## [49] processx_3.6.1 tidygraph_1.2.1 rlang_1.0.3
## [52] zeallot_0.1.0 systemfonts_1.0.4 GlobalOptions_0.1.2
## [55] splines_4.2.1 lazyeval_0.2.2 broom_1.0.0
## [58] BiocManager_1.30.18 yaml_2.3.5 reshape2_1.4.4
## [61] modelr_0.1.8 backports_1.4.1 qvalue_2.28.0
## [64] usethis_2.1.6 tools_4.2.1 bookdown_0.27
## [67] ggplotify_0.1.0 ellipsis_0.3.2 jquerylib_0.1.4
## [70] RColorBrewer_1.1-3 proxy_0.4-27 BiocGenerics_0.42.0
## [73] sessioninfo_1.2.2 Rcpp_1.0.8.3 plyr_1.8.7
## [76] progress_1.2.2 base64enc_0.1-3 zlibbioc_1.42.0
## [79] RCurl_1.98-1.7 ps_1.7.1 prettyunits_1.1.1
## [82] GetoptLong_1.0.5 viridis_0.6.2 S4Vectors_0.34.0
## [85] haven_2.5.0 ggrepel_0.9.1 cluster_2.1.3
## [88] here_1.0.1 fs_1.5.2 magrittr_2.0.3
## [91] data.table_1.14.2 DO.db_2.9 circlize_0.4.15
## [94] reprex_2.0.1 whisker_0.4 ggnewscale_0.4.7
## [97] matrixStats_0.62.0 pkgload_1.3.0 hms_1.1.1
## [100] patchwork_1.1.1 evaluate_0.15 readxl_1.4.0
## [103] IRanges_2.30.0 gridExtra_2.3 shape_1.4.6
## [106] tfruns_1.5.0 compiler_4.2.1 crayon_1.5.1
## [109] shadowtext_0.1.2 htmltools_0.5.2 ggfun_0.0.6
## [112] tzdb_0.3.0 aplot_0.1.6 lubridate_1.8.0
## [115] DBI_1.1.3 tweenr_1.0.2 dbplyr_2.2.1
## [118] MASS_7.3-57 Matrix_1.4-1 cli_3.3.0
## [121] parallel_4.2.1 igraph_1.3.2 pkgconfig_2.0.3
## [124] pkgdown_2.0.5 xml2_1.3.3 foreach_1.5.2
## [127] ggtree_3.4.0 bslib_0.3.1 rngtools_1.5.2
## [130] XVector_0.36.0 doFuture_0.12.2 rvest_1.0.2
## [133] doRNG_1.8.2 yulab.utils_0.0.5 callr_3.7.0
## [136] digest_0.6.29 Biostrings_2.64.0 rmarkdown_2.14
## [139] cellranger_1.1.0 fastmatch_1.1-3 tidytree_0.3.9
## [142] nloptr_2.0.3 rjson_0.2.21 lifecycle_1.0.1
## [145] nlme_3.1-157 jsonlite_1.8.0 desc_1.4.1
## [148] viridisLite_0.4.0 fansi_1.0.3 pillar_1.7.0
## [151] lattice_0.20-45 KEGGREST_1.36.2 fastmap_1.1.0
## [154] httr_1.4.3 pkgbuild_1.3.1 GO.db_3.15.0
## [157] remotes_2.4.2 glue_1.6.2 png_0.1-7
## [160] iterators_1.0.14 bit_4.0.4 ggforce_0.3.3
## [163] stringi_1.7.6 sass_0.4.1 blob_1.2.3
## [166] textshaping_0.3.6 memoise_2.0.1 ape_5.6-2